

Los científicos están desentrañando el misterio de qué desencadena la enfermedad de Huntington, un trastorno hereditario devastador y fatal que ataca en la flor de la vida, provocando que las células nerviosas en partes del cerebro se descompongan y mueran.
La mutación genética asociada a la enfermedad de Huntington se conoce desde hace mucho tiempo, pero los científicos no han entendido cómo las personas pueden tener la mutación desde el nacimiento y no desarrollar ningún problema hasta más tarde en la vida.
Una nueva investigación muestra que la mutación es, sorprendentemente, inofensiva durante décadas, pero que va creciendo silenciosamente hasta convertirse en una mutación mayor, hasta que finalmente cruza un umbral, genera proteínas tóxicas y mata las células en las que se ha expandido.
“El dilema en nuestro campo ha sido: ¿por qué se tiene un trastorno genético que se manifiesta más tarde en la vida si el gen está presente en el momento de la concepción?”, dijo el Dr. Mark Mehler, quien dirige el Instituto de Trastornos Cerebrales y Regeneración Neural en el Colegio de Medicina Albert Einstein y no participó en la investigación. Calificó la investigación como un estudio “histórico” y dijo que “aborda muchos de los problemas que han plagado el campo durante mucho tiempo”.
La muerte de las células cerebrales acaba provocando problemas de movimiento, pensamiento y comportamiento. Los síntomas de Huntington (que incluyen movimientos involuntarios, marcha inestable, cambios de personalidad y deterioro del juicio) suelen comenzar entre los 30 y los 50 años y empeoran gradualmente a lo largo de 10 a 25 años.
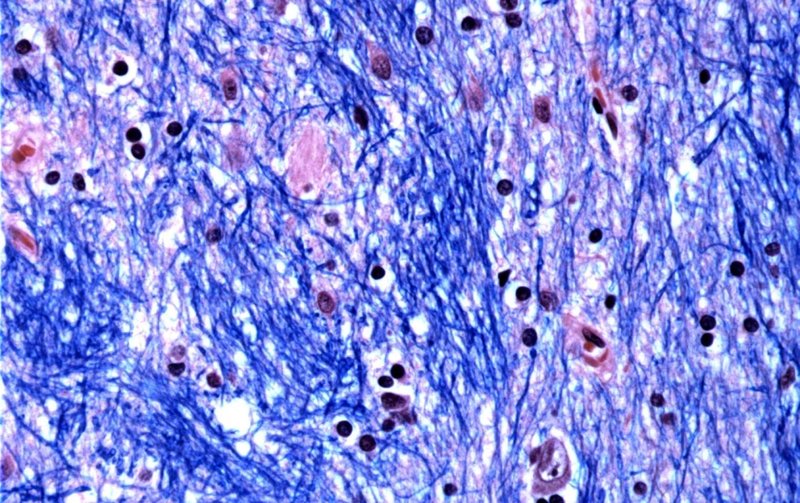
Científicos del Instituto Broad del MIT y Harvard, del Hospital McLean de Massachusetts y de la Escuela de Medicina de Harvard estudiaron tejido cerebral donado por 53 personas con Huntington y 50 sin ella, analizando medio millón de células.
Se centraron en la mutación de Huntington, que implica un tramo de ADN en un gen particular donde una secuencia de tres letras –CAG– se repite al menos 40 veces. En las personas sin la enfermedad, esta secuencia se repite solo entre 15 y 35 veces. Descubrieron que los tramos de ADN con 40 o más de esas “repeticiones” se expanden con el tiempo hasta tener cientos de CAG de longitud. Una vez que las CAG alcanzan un umbral de aproximadamente 150, ciertos tipos de neuronas enferman y mueren.
Los hallazgos “fueron realmente sorprendentes, incluso para nosotros”, dijo Steve McCarroll, miembro de Broad y coautor principal del estudio, que fue publicado el jueves en la revista Cell. El estudio fue financiado en parte por el Instituto Médico Howard Hughes, una organización que también apoya al departamento de Salud y Ciencia de The Associated Press.
El equipo de investigación estimó que los tractos repetidos crecen lentamente durante las primeras dos décadas de vida y luego el ritmo se acelera dramáticamente cuando alcanzan alrededor de 80 CAG.
«Cuanto más largas sean las repeticiones, más temprano en la vida se producirá el inicio», dijo la investigadora en neurociencia Sabina Berretta, una de las autoras principales del estudio.
Los investigadores reconocieron que algunos científicos se mostraron inicialmente escépticos cuando se compartieron los resultados en conferencias, ya que trabajos anteriores habían descubierto que las expansiones repetidas en el rango de 30 a 100 CAG eran necesarias, pero no suficientes, para causar la enfermedad de Huntington. McCarroll estuvo de acuerdo en que 100 CAG o menos no son suficientes para desencadenar la enfermedad, pero dijo que su estudio descubrió que las expansiones con al menos 150 CAG sí lo son.
Los investigadores esperan que sus hallazgos puedan ayudar a los científicos a encontrar formas de retrasar o prevenir esta enfermedad incurable, que afecta a unos 41.000 estadounidenses y que ahora se trata con medicamentos para controlar los síntomas.
Recientemente, los ensayos con medicamentos experimentales diseñados para reducir los niveles de la proteína producida por el gen de Huntington mutado han tenido dificultades. Los nuevos hallazgos sugieren que esto se debe a que pocas células tienen la versión tóxica de la proteína en un momento dado.
Los investigadores dijeron que retardar o detener la expansión de las repeticiones de ADN puede ser una mejor manera de atacar la enfermedad.
Aunque no hay garantías de que esto evite la enfermedad de Huntington, McCarroll dijo que «muchas empresas están iniciando o ampliando programas para intentar lograrlo».




{kind=link}